Orchestrating Self-Replication in Artificial Cells through Digital Microfluidics
10.1101/2025.05.23.655734

DMF Replicator
A defining feature of living cells is their ability to self-replicate; but creating artificial cells with this capability remains challenging, due to the complexity of biological division machinery. Rather than seeking to reconstitute this machinery, here we take direct control of DNA replication and compartment division using digital microfluidics. This approach allows us to precisely orchestrate these two fundamental processes, providing insight into how they must be coupled for successful self-replication.
Triply Responsive Control of Anion/Cation Transport with an Artificial Ion Channel Responsive to Multiple Stimuli
10.26434/chemrxiv-2025-sbp1x

azo-p5 ion channels.
We present a synthetic ion channel whose activity can be controlled by three orthogonal stimuli (light, pH, guest/ligand). The channel is formed from a pillar[5]arene functionalized with photoswitchable tetrafluoroazobenzene moieties. We demonstrate excellent control over E:Z switching across ten incorporated photoswitches (E to Z 87%, Z to E quant.). We show that the most active isomer – the Z-isomer – forms dimeric ion channels in membranes, with selectivity for M+/Cl− symport. Single molecule planar bilayer conductance studies show distinct high and low conductance states dependent on irradiation wavelength. Finally, we demonstrate that this activity can be modulated over 170-fold by controlling pH, irradiation, and guest addition, creating a powerful addition to the canon of synthetic ion channels.
Rational Design Principles for De Novo α-Helical Peptide Barrels with Dynamic Conductive Channels
10.1021/jacs.4c13933

α-helical peptide channels.
Despite advances in peptide and protein design, the rational design of membrane-spanning peptides that form conducting channels remains challenging due to our imperfect understanding of the sequence-to-structure relationships that drive membrane insertion, assembly, and conductance. Here, we describe the design and computational and experimental characterization of a series of coiled coil-based peptides that form transmembrane α-helical barrels with conductive channels.
Real-time label-free imaging of living crystallization-driven self-assembly
10.1038/s41467-025-57776-9
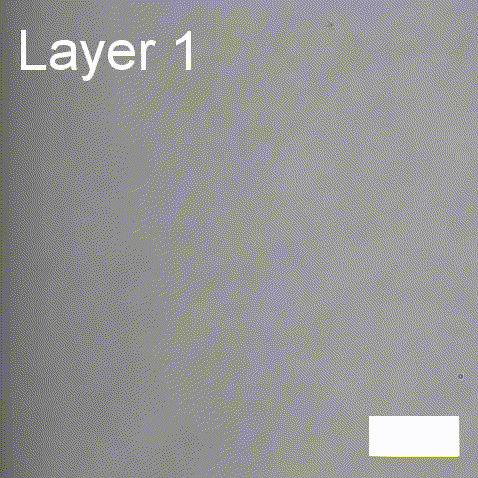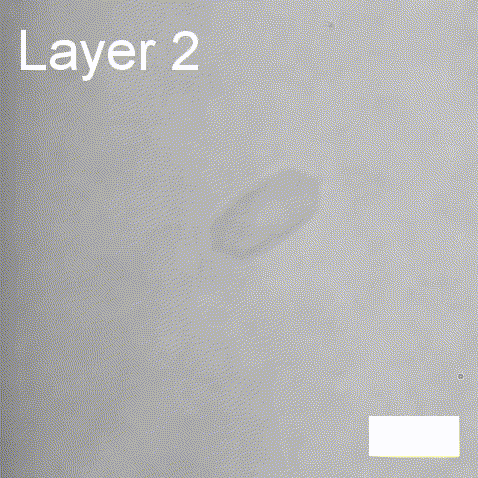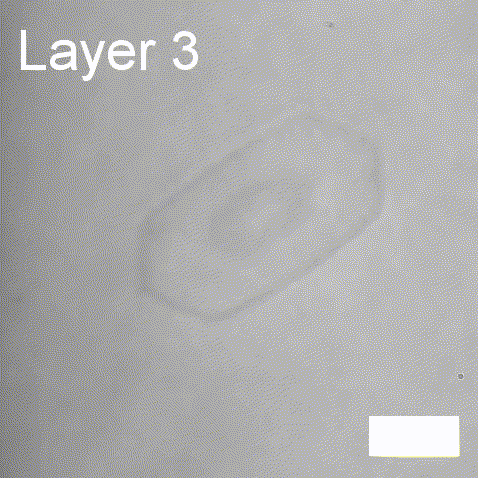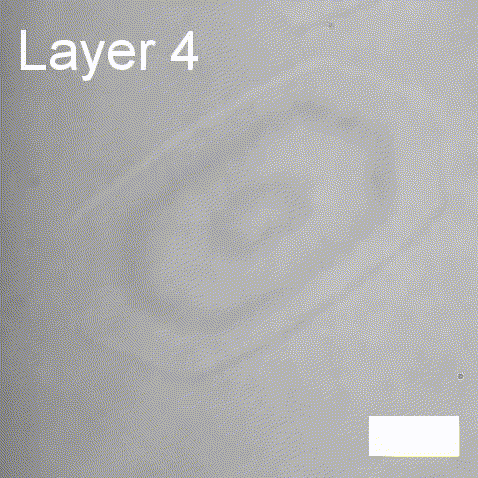
Real-time label-free imaging of living crystallization-driven self-assembly.
Our latest work now published in Nature Communications. This is the last of our pandemic papers, so no membrane biophysics! Applying label-free microscopy to watch polymer self-assembly in real-time. This lets us map directly how reaction parameters affect the composition and growth of individual 2D polymer microplatelets. A fantastic collaboration with Rachel O’Reilly at the University of Birmingham. Congratulations go to Yujie Guo for another fantastic bit of science.
Ion Transport and Membrane Channel Formation Using a Peptidomimetic in Droplet Interface Bilayers
10.1039/D4CC05926C

Peptidomimetic channels.
Herein, we present a synthetic peptidomimetic, TBP2, which forms artificial membrane channels within Droplet Interface Bilayers (DIBs). Through real-time electrophysiology, TIRF microscopy, and single-channel recordings, we demonstrate the ability of TBP2 to mediate high-conductance transport of Na⁺ and K⁺ across the DIBs, highlighting its potential for
nanobiotechnology applications in cell-free systems.
α-helical peptide channels.
Despite advances in peptide and protein design, the rational design of membrane-spanning peptides that form conducting channels remains challenging due to our imperfect understanding of the sequence-to-structure relationships that drive membrane insertion, assembly, and conductance. Here, we describe the design and computational and experimental characterization of a series of coiled coil-based peptides that form transmembrane α-helical barrels.

Spatial light modulation for interferometric scattering microscopy
.
Interferometric scattering (iSCAT) microscopy enables high-speed and label-free detection of individual molecules and small nanoparticles. Here we apply point spread function engineering to provide adaptive control of iSCAT images using spatial light modulation. With this approach, we demonstrate improved dynamic spatial filtering, real-time background subtraction, focus control, and signal modulation based on sample orientation.

Monitoring AuNP growth.
Methods capable of controlling synthesis at the level of an individual nanoparticle are a key step towards improved reproducibility and scalability in engineering complex nanomaterials. To address this we combine spatially-patterned activation of the photoreductant sodium pyruvate with interferometric scattering microscopy to achieve fast, label-free monitoring and control of hundreds of gold nanoparticles in real-time. Individual particle growth kinetics are well-described by two-step nucleation autocatalysis model, but with a distribution of individual rate constants that changes with reaction conditions.
Spatiotemporal stop-and-go dynamics of the mitochondrial TOM core complex correlates with channel activity
10.1038/s42003-022-03419-4
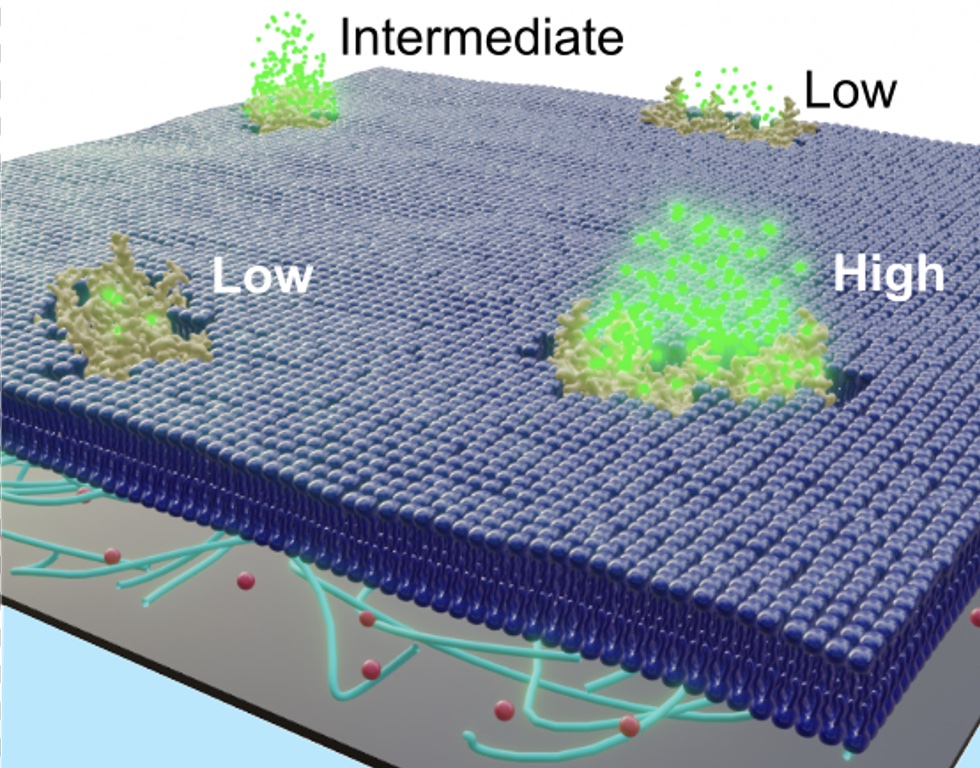
TOM diffusion is coupled to permeability.
Here we show the correlation between lateral protein diffusion and channel activity of the general protein import pore of mitochondria (TOM-CC) in membranes resting on ultrathin hydrogel films. Using electrode-free optical recordings of ion flux, we find that TOM-CC switches reversibly between three states of ion permeability associated with protein diffusion.
While freely diffusing TOM-CC molecules are predominantly in a high permeability state, non-mobile molecules are mostly in an intermediate or low permeability state. We explain this behavior by the mechanical binding of the two protruding Tom22 subunits to the hydrogel and a concomitant combinatorial opening and closing of the two β-barrel pores of TOM-CC. TOM-CC could thus represent a β-barrel membrane protein complex to exhibit membrane state-dependent mechanosensitive properties, mediated by its two Tom22 subunits.
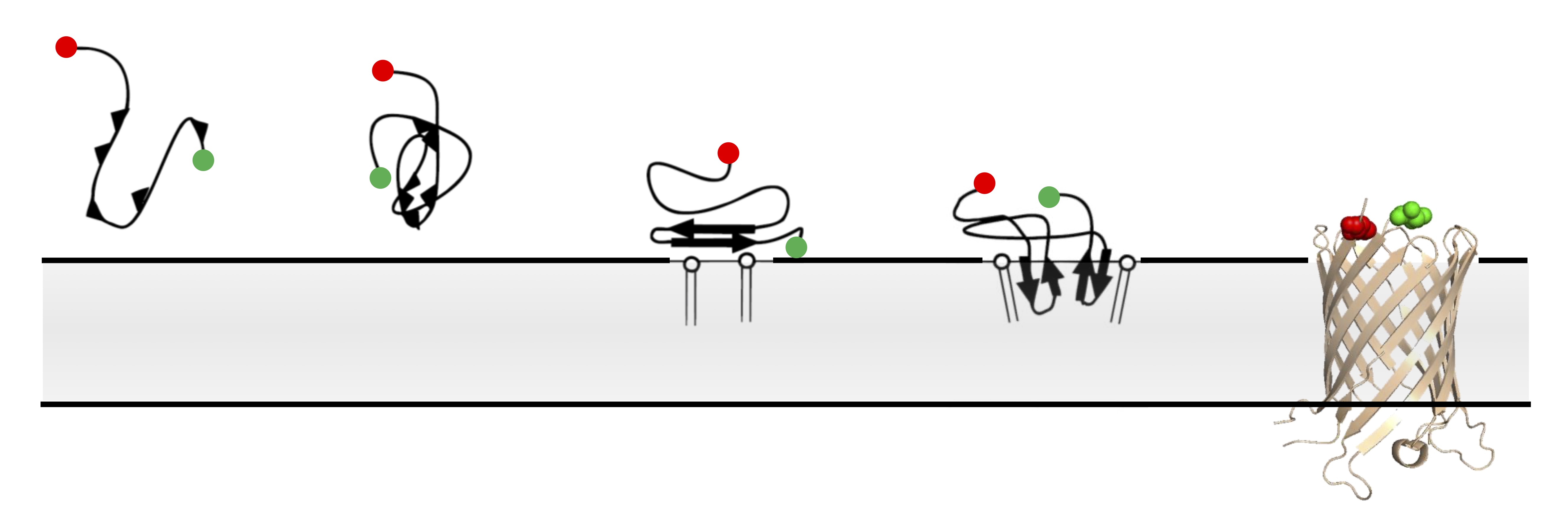
Single-molecule FRET imaging of beta-barrel folding.
In comparison to globular proteins, the spontaneous folding and insertion of β-barrel membrane proteins is surprisingly slow, typically occurring on the order of minutes. Using single-molecule Förster Resonance Energy Transfer to report on the folding of fluorescently-labelled Outer Membrane Protein G we measured the real-time insertion of a β-barrel membrane protein from an unfolded state. Folding events were rare, and fast (<20 ms); occurring immediately upon arrival at the membrane. This combination of infrequent, but rare, folding resolves this apparent dichotomy between slow ensemble kinetics, and the typical timescales of biomolecular folding.
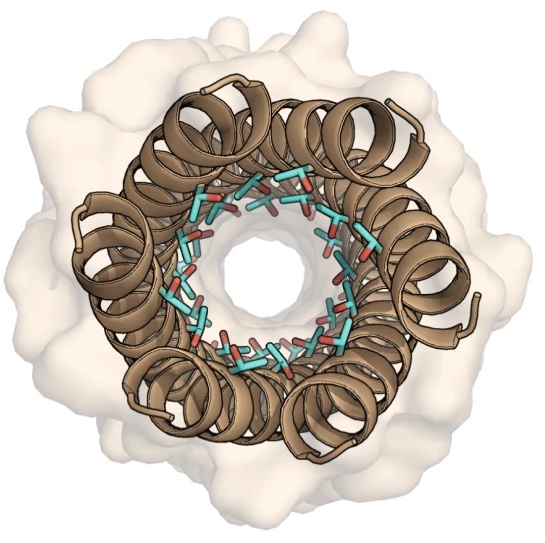
Designer alpha-helical barrels.
The design of peptides that assemble in membranes to form functional ion channels is challenging. Specifically, hydrophobic interactions must be designed between the peptides and at the peptide–lipid interfaces simultaneously. Here, we take a multi-step approach towards this problem. First, we use rational de novo design to generate water-soluble α-helical barrels with polar interiors, and confirm their structures using high-resolution X-ray crystallography. These α-helical barrels have water-filled lumens like those of transmembrane channels. Next, we modify the sequences to facilitate their insertion into lipid bilayers. Single-channel electrical recordings and fluorescent imaging of the peptides in membranes show monodisperse, cation-selective channels of unitary conductance.
Single-molecule imaging of pore-forming toxin dynamics in droplet interface bilayers
10.1016/bs.mie.2021.01.035

Our chapter in Methods 619.
Single-channel recording from pore-forming toxins (PFTs) provides a clear and direct molecular readout of toxin action. However to complete any mechanistic understanding of PFT behavior, this functional kinetic readout must be linked to the underlying changes in toxin structure, binding, conformation, or stoichiometry. Here we review how single-molecule imaging methods might be used to further our understanding of PFTs, and provide detailed practical guidance on the use of droplet interface bilayers as a method capable of examining both single-molecule fluorescence and single-channel electrical signals from PFTs.

Tracking Perfingolysin O assembly and pore formation.
We exploit single-molecule tracking and optical single channel recording in droplet interface bilayers to resolve the assembly pathway of the Cholesterol-Dependent Cytolysin, Perfringolysin O. This enables quantification of the stoichiometry of PFO complexes during assembly with millisecond temporal resolution and 20 nanometre spatial precision. Our results support a model of overall stepwise irreversible assembly, dominated by monomer addition, but with infrequent assembly from larger partial complexes. Furthermore, our results suggest a dominant proportion of inserted, but non-conductive intermediates in assembly.

Artificial Signal Transduction across Membranes. A key conundrum in the construction of an artificial cell is to simultaneously maintain a robust physical barrier to the external environment, while also providing efficient exchange of information across this barrier. Biomimicry provides a number of avenues by which such requirements might be met. Herein, we provide a brief introduction to the challenges facing this field and explore progress to date.
Modifying membrane morphology and interactions with DNA origami clathrin-mimic networks
10.1021/acsnano.8b07734

We designed a three-arm DNA origami nanostructure whose shape resembles that of the clathrin triskelion.
We describe the triggered assembly of a bioinspired DNA origami meshwork on a lipid membrane. DNA triskelia, three-armed DNA origami nanostructures inspired by the membrane-modifying protein clathrin, are bound to lipid mono- and bilayers using cholesterol anchors. Polymerization of triskelia, triggered by the addition of DNA staples, links triskelion arms to form a mesh. Using transmission electron microscopy, we observe nanoscale local deformation of a lipid monolayer induced by triskelion polymerization that is reminiscent of the formation of clathrin-coated pits. We also show that the polymerization of triskelia bound to lipid bilayers modifies interactions between them, inhibiting the formation of a synapse between giant unilamellar vesicles and a supported lipid bilayer.
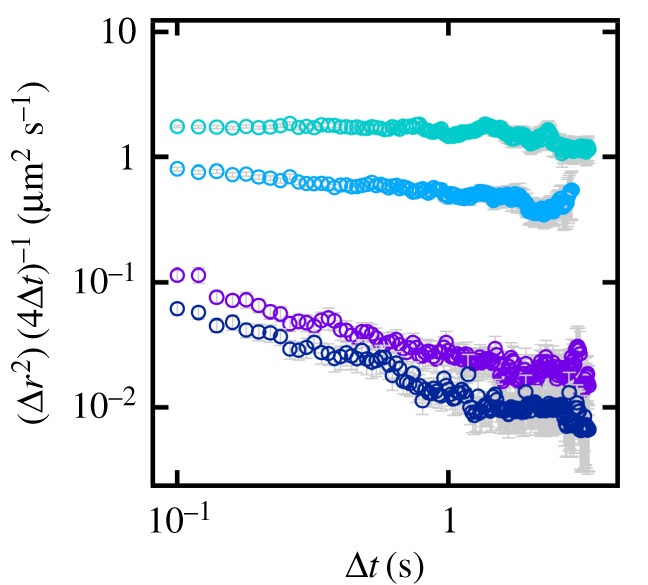
Anomalous diffusion increases over time from 15 s (turquoise) to 10 min (dark blue).
Diffusion in biological membranes is seldom simply Brownian motion; instead, the rate of diffusion is dependent on the time scale of observation and so is often described as anomalous. In order to help better understand this phenomenon, model systems are needed where the anomalous diffusion of the lipid bilayer can be tuned and quantified. We recently demonstrated one such model by controlling the excluded area fraction in supported lipid bilayers (SLBs) through the incorporation of lipids derivatized with polyethylene glycol. Here, we extend this work, using urea to induce anomalous diffusion in SLBs. By tuning incubation time and urea concentration, we produce bilayers that exhibit anomalous behaviour on the same scale as that observed in biological membranes.
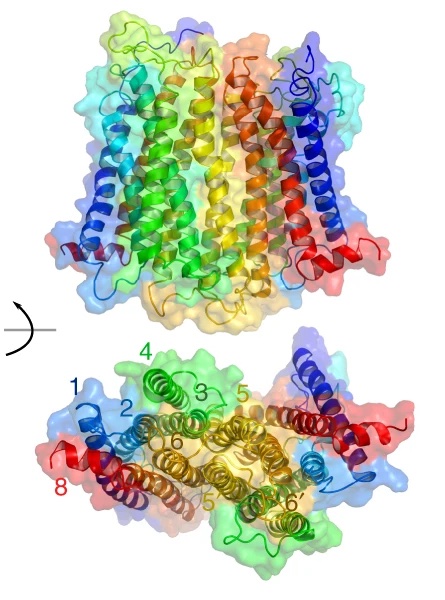
Proposed oligomerisation interfaces for NTS1 .
G protein-coupled receptors (GPCRs) are the largest class of membrane receptors, playing a key role in the regulation of processes as varied as neurotransmission and immune response. Evidence for GPCR oligomerisation has been accumulating that challenges the idea that GPCRs function solely as monomeric receptors; however, GPCR oligomerisation remains controversial primarily due to the difficulties in comparing evidence from very different types of structural and dynamic data. Using a combination of single-molecule and ensemble FRET, double electron–electron resonance spectroscopy, and simulations, we show that dimerisation of the GPCR neurotensin receptor 1 is regulated by receptor density and is dynamically tuneable over the physiological range. We propose a “rolling dimer” interface model in which multiple dimer conformations co-exist and interconvert. These findings unite previous seemingly conflicting observations, provide a compelling mechanism for regulating receptor signalling, and act as a guide for future physiological studies.
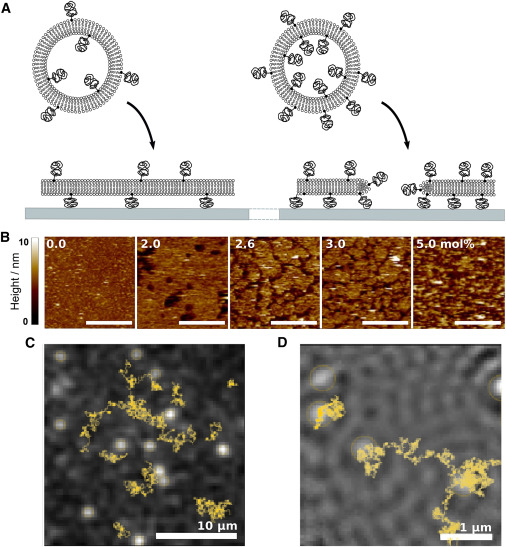
As the mole fraction of PEGylated lipids increases, defects form in the bilayer that act as obstacles, generating anomalous diffusion.
Diffusion in cell membranes is not just simple two-dimensional Brownian motion but typically depends on the timescale of the observation. The physical origins of this anomalous subdiffusion are unresolved, and model systems capable of quantitative and reproducible control of membrane diffusion have been recognized as a key experimental bottleneck. Here, we control anomalous diffusion using supported lipid bilayers containing lipids derivatized with polyethylene glycol (PEG) headgroups. Bilayers with specific excluded area fractions are formed by control of PEG lipid mole fraction. These bilayers exhibit a switch in diffusive behavior, becoming anomalous as bilayer continuity is disrupted. Using a combination of single-molecule fluorescence and interferometric imaging, we measure the anomalous behavior in this model over four orders of magnitude in time. Diffusion in these bilayers is well described by a power-law dependence of the mean-square displacement with observation time. Anomaleity in this system can be tailored by simply controlling the mole fraction of PEG lipid, producing bilayers with diffusion parameters similar to those observed for anomalous diffusion in biological membranes.
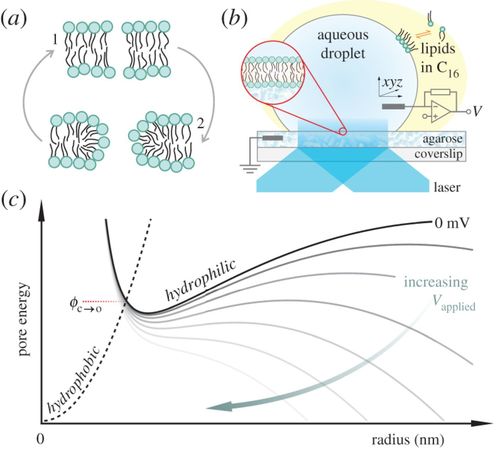
A hydrophobic pore that surmounts the energy barrier will convert to a toroidal pore. The applied transmembrane potential lowers the pore free energy.
Electroporation is a common tool for gene transfection, tumour ablation, sterilization and drug delivery. Using experimental methods, we explore the temperature dependence of electropore formation in a model membrane system (droplet-interface bilayers), using optical single-channel recording to image the real-time gating of individual electropores. We investigate the influence of the agarose substrate on electropores formed in this system. Furthermore, by examining the temperature-dependent kinetics of pore opening and closure we are able to estimate a barrier to pore opening in 1,2-diphytanoyl-sn-glycero-3-phosphocholine (DPhPC) membranes to be 25.0 ± 8.3 kT, in agreement with previous predictions. Overall these measurements help support the toroidal model of membrane electroporation.
This article is part of the themed issue ‘Membrane pores: from structure and assembly, to medicine and technology’.
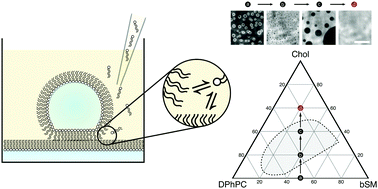
Control of membrane composition using Droplet Interface Bilayers.
Changes in local lipid composition are thought to play a key role in regulating many complex cellular processes. By studying lipid organization in artificial lipid bilayers the physical principles underlying these process can be studied in detail. However, such in vitro measurements are often hindered by heterogeneities in the lipid composition of individual bilayers prepared by current bulk methods. Here, the lipid composition of an individual droplet interface bilayer is varied by lipid titration into the bilayer from the oil phase in a microfluidic device. Control of lipid composition allows the reversible switching between single- and two-phase regions and sampling of specific lipid compositions in an individual bilayer. This method enables controlled modulation of composition-sensitive processes in a single lipid membrane.

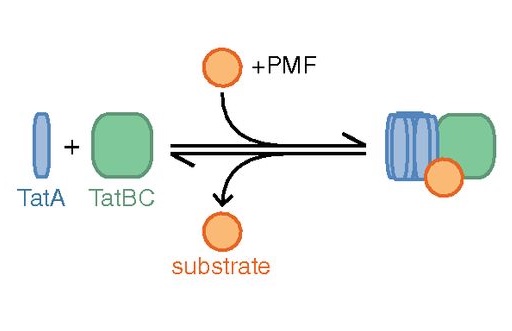
Fluorescence images of TatA-YFP complexes in living cells.
The twin-arginine protein translocation system (Tat) transports folded proteins across the bacterial cytoplasmic membrane and the thylakoid membranes of plant chloroplasts. The Tat transporter is assembled from multiple copies of the membrane proteins TatA, TatB, and TatC. We combine sequence co-evolution analysis, molecular simulations, and experimentation to define the interactions between the Tat proteins of Escherichia coli at molecular-level resolution. In the TatBC receptor complex the transmembrane helix of each TatB molecule is sandwiched between two TatC molecules, with one of the inter-subunit interfaces incorporating a functionally important cluster of interacting polar residues. Unexpectedly, we find that TatA also associates with TatC at the polar cluster site. Our data provide a structural model for assembly of the active Tat translocase in which substrate binding triggers replacement of TatB by TatA at the polar cluster site. Our work demonstrates the power of co-evolution analysis to predict protein interfaces in multi-subunit complexes.

Elecropore artwork.
Electroporation is a widely used technique to permeabilize cell membranes. Despite its prevalence, our understanding of the mechanism of voltage-mediated pore formation is incomplete; methods capable of visualizing the time-dependent behavior of individual electropores would help improve our understanding of this process. Here, using optical single-channel recording, we track multiple isolated electropores in real time in planar droplet interface bilayers. We observe individual, mobile defects that fluctuate in size, exhibiting a range of dynamic behaviors. We observe fast (25 s^−1) and slow (2 s^−1) components in the gating of small electropores, with no apparent dependence on the applied potential. Furthermore, we find that electropores form preferentially in the liquid disordered phase. Our observations are in general supportive of the hydrophilic toroidal pore model of electroporation, but also reveal additional complexity in the interactions, dynamics, and energetics of electropores.
Chemical amplification of magnetic field effects relevant to avian magnetoreception.
10.1038/nchem.2447.
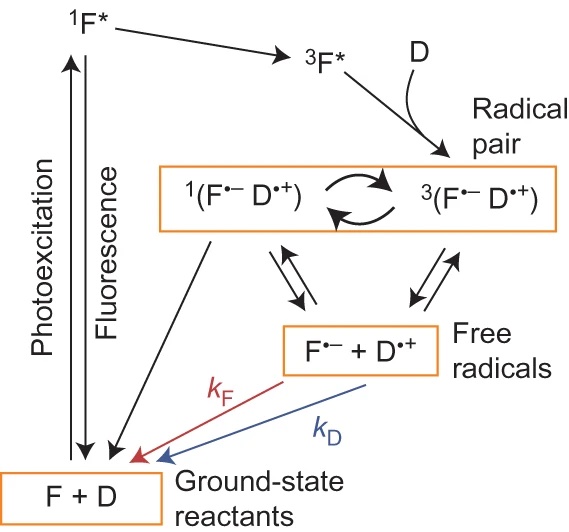
Reaction scheme for the intermolecular photochemical reaction of a flavin (F) with an electron donor (D).
Magnetic fields as weak as the Earth’s can change the yields of radical pair reactions even though the energies involved are orders of magnitude smaller than the thermal energy, kBT, at room temperature. Proposed as the source of the light-dependent magnetic compass in migratory birds, the radical pair mechanism is thought to operate in cryptochrome flavoproteins in the retina. Here we demonstrate that the primary magnetic field effect on flavin photoreactions can be amplified chemically by slow radical termination reactions under conditions of continuous photoexcitation. The nature and origin of the amplification are revealed by studies of the intermolecular flavin–tryptophan and flavin–ascorbic acid photocycles and the closely related intramolecular flavin–tryptophan radical pair in cryptochrome. Amplification factors of up to 5.6 were observed for magnetic fields weaker than 1 mT. Substantial chemical amplification could have a significant impact on the viability of a cryptochrome-based magnetic compass sensor.
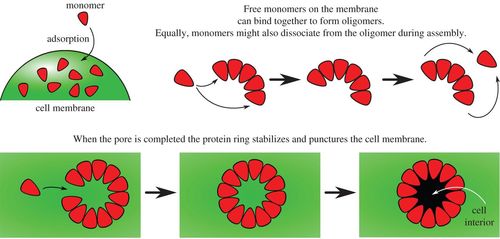
Schematic diagrams illustrating monomer addition as a proposed pathway to pore formation.
Pore-forming toxins are ubiquitous cytotoxins that are exploited by both bacteria and the immune response of eukaryotes. These toxins kill cells by assembling large multimeric pores on the cell membrane. However, a quantitative understanding of the mechanism and kinetics of this self-assembly process is lacking. We propose an analytically solvable kinetic model for stepwise, reversible oligomerization. In biologically relevant limits, we obtain simple algebraic expressions for the rate of pore formation, as well as for the concentration of pores as a function of time. Quantitative agreement is obtained between our model and time-resolved kinetic experiments of Bacillus thuringiensis Cry1Ac (tetrameric pore), aerolysin, Staphylococcus aureus α-haemolysin (heptameric pores) and Escherichia coli cytolysin A (dodecameric pore). Furthermore, our model explains how rapid self-assembly can take place with low concentrations of oligomeric intermediates, as observed in recent single-molecule fluorescence experiments of α-haemolysin self-assembly. We propose that suppressing the concentration of oligomeric intermediates may be the key to reliable, error-free, self-assembly of pores.
Length-Dependent Formation of Transmembrane Pores by 310-Helical α-Aminoisobutyric Acid Foldamers
10.1021/jacs.5b12057
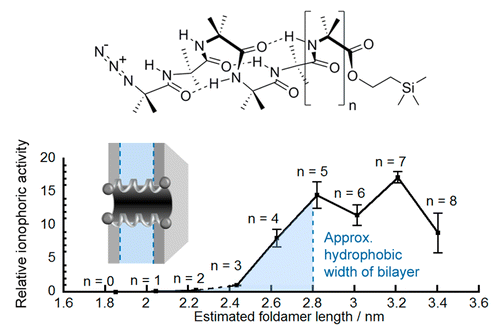
Foldamer designs to match membrane thickness.
The synthetic biology toolbox lacks extendable and conformationally controllable yet easy-to-synthesize building blocks that are long enough to span membranes. To meet this need, an iterative synthesis of α-aminoisobutyric acid (Aib) oligomers was used to create a library of homologous rigid-rod 310-helical foldamers, which have incrementally increasing lengths and functionalizable N- and C-termini. This library was used to probe the inter-relationship of foldamer length, self-association strength, and ionophoric ability, which is poorly understood. Although foldamer self-association in nonpolar chloroform increased with length, with a ∼14-fold increase in dimerization constant from Aib6 to Aib11, ionophoric activity in bilayers showed a stronger length dependence, with the observed rate constant for Aib11 ∼70-fold greater than that of Aib6. The strongest ionophoric activity was observed for foldamers with >10 Aib residues, which have end-to-end distances greater than the hydrophobic width of the bilayers used (∼2.8 nm); X-ray crystallography showed that Aib11 is 2.93 nm long. These studies suggest that being long enough to span the membrane is more important for good ionophoric activity than strong self-association in the bilayer. Planar bilayer conductance measurements showed that Aib11 and Aib13, but not Aib7, could form pores. This pore-forming behavior is strong evidence that Aibm (m ≥ 10) building blocks can span bilayers.
The TatC component of the twin-arginine protein translocase functions as an obligate oligomer.
10.1111/mmi.13106
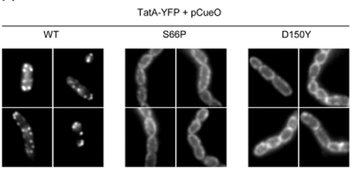
Substrate-induced assembly of TatA-YFP is blocked by TatC mutations.
The Tat protein export system translocates folded proteins across the bacterial cytoplasmic membrane and the plant thylakoid membrane. The Tat system in Escherichia coli is comprised of TatA, TatB and TatC proteins. TatB and TatC form an oligomeric, multivalent receptor complex that binds Tat substrates, while multiple protomers of TatA assemble at substrate-bound TatBC receptors to facilitate substrate transport. We have addressed whether oligomerisation of TatC is an absolute requirement for operation of the Tat pathway by screening for dominant negative alleles of tatC that inactivate Tat function in the presence of wild type tatC. Single substitutions that confer dominant negative TatC activity localised to the periplasmic cap region. The variant TatC proteins retained the ability to interact with TatB and with a Tat substrate but were unable to support the in vivo assembly of TatA complexes. Blue-native PAGE analysis showed that the variant TatC proteins produced smaller TatBC complexes than the wild type TatC protein. The substitutions did not alter disulphide crosslinking to neighbouring TatC molecules from positions in the periplasmic cap but abolished a substrate-induced disulphide crosslink in transmembrane helix five of TatC. Our findings show that TatC functions as an obligate oligomer.
Imaging potassium-flux through individual electropores in droplet interface bilayers.
10.1016/j.bbamem.2015.07.009
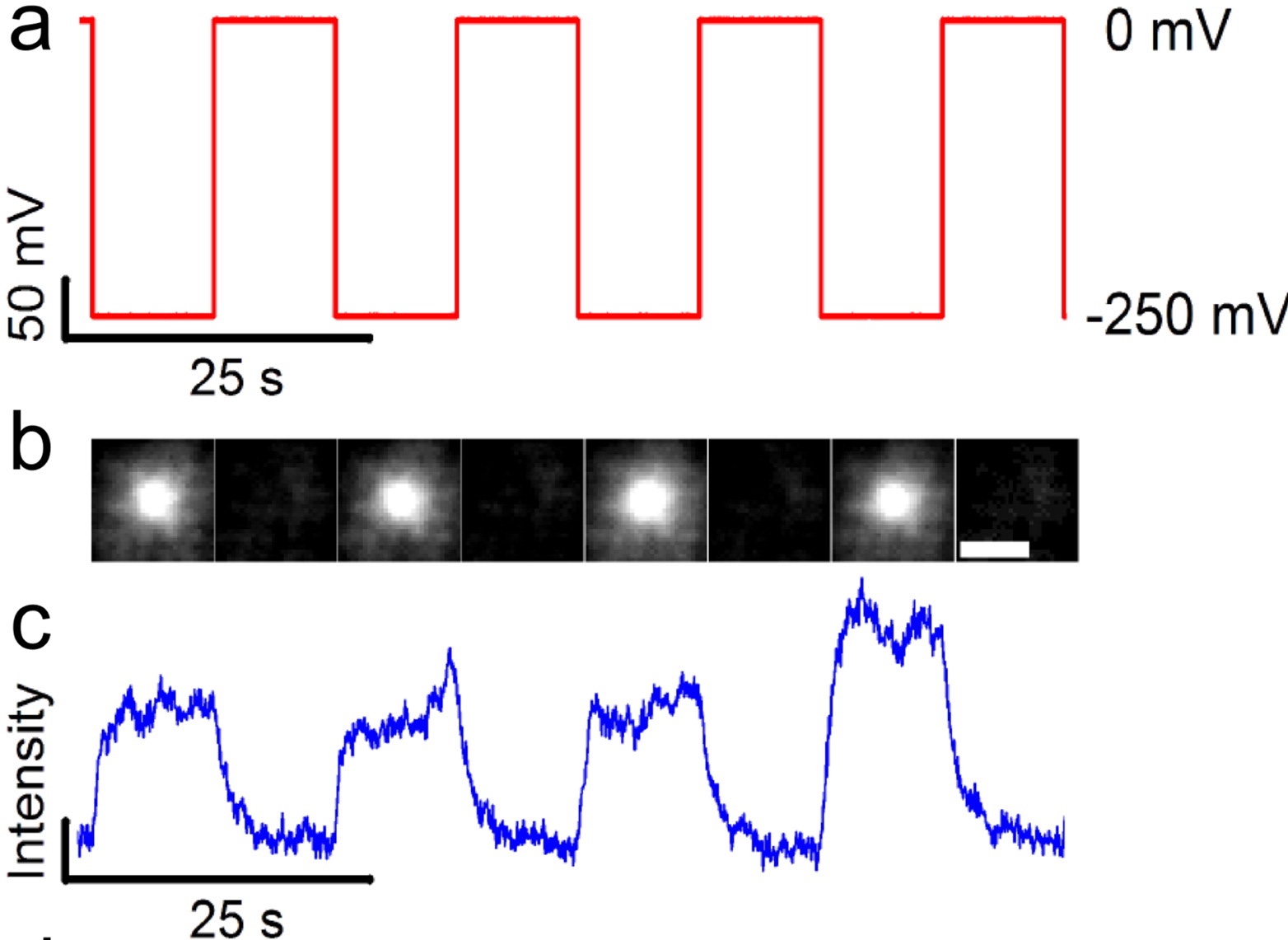
Optical measurement of potassium flux through a representative pore.
Using total internal reflection fluorescence microscopy of droplet interface bilayers containing the potassium-sensitive fluorophore APG-4, we imaged the ionic flux through individual electropores. We are able to monitor up to 30 individual pores in parallel and show voltage dependent responses in fluorescence that corresponds to the measured ionic current. These experiments help quantify the scope and current limitations of optical single channel recordings of potassium flux.

RNA sensing by optical Single Channel Recording.
Protein nanopores such as α-haemolysin and Mycobacterium smegmatis porin A (MspA) can be used to sequence long strands of DNA at low cost. To provide high-speed sequencing, large arrays of nanopores are required, but current nanopore sequencing methods rely on ionic current measurements from individually addressed pores and such methods are likely to prove difficult to scale up. Here we show that, by optically encoding the ionic flux through protein nanopores, the discrimination of nucleic acid sequences and the detection of sequence-specific nucleic acid hybridization events can be parallelized. We make optical recordings at a density of ∼104 nanopores per mm2 in a single droplet interface bilayer. Nanopore blockades can discriminate between DNAs with sub-picoampere equivalent resolution, and specific miRNA sequences can be identified by differences in unzipping kinetics. By creating an array of 2,500 bilayers with a micropatterned hydrogel chip, we are also able to load different samples into specific bilayers suitable for high-throughput nanopore recording.

The nanodomain lifetimes we measure are on a similar order to the kinetics reported for many membrane-signaling processes.
Lipid rafts are submicron proteolipid domains thought to be responsible for membrane trafficking and signaling. Their small size and transient nature put an understanding of their dynamics beyond the reach of existing techniques, leading to much contention as to their exact role. Here, we exploit the differences in light scattering from lipid bilayer phases to achieve dynamic imaging of nanoscopic lipid domains without any labels. Using phase-separated droplet interface bilayers we resolve the diffusion of domains as small as 50 nm in radius and observe nanodomain formation, destruction, and dynamic coalescence with a domain lifetime of 220 ± 60 ms. Domain dynamics on this timescale suggests an important role in modulating membrane protein function.
Probing channel, pump, and transporter function using single-molecule fluorescence.
10.1002/9781119085126.ch11
![]()
Our chapter in Pumps, Channels, and Transporters.
The ability of patch clamping to monitor individual ion channels revolutionized our approach to understanding the discrete functional states of these membrane proteins, including how ligands, point mutations, and the electrochemical potential affect channel function.
Fluorescence-detected magnetic field effects on radical pair reactions from femtolitre volumes.
10.1039/C5CC01099CDod
Detection of Magnetic Field Effects in radical pair reactions from 100 femtolitres.
We show that the effects of applied magnetic fields on radical pair reactions can be sensitively measured from sample volumes as low as ∼100 femtolitres using total internal reflection fluorescence microscopy. Development of a fluorescence-based microscope method is likely to be a key step in further miniaturisation that will allow detection of magnetic field effects on single molecules.

Detection of Magnetic Field Effects in radical pair reactions from 100 femtolitres.
Pore-forming proteins (PFPs) interact with lipid bilayers to compromise membrane integrity. Many PFPs function by inserting a ring of oligomerized subunits into the bilayer to form a protein-lined hydrophilic channel. However, mounting evidence suggests that PFPs can also generate ‘proteolipidic’ pores by contributing to the fusion of inner and outer bilayer leaflets to form a toroidal structure. We discuss here toroidal pore formation by peptides including melittin, protegrin, and Alzheimer’s Aβ1-41, as well as by PFPs from several evolutionarily unrelated families: the colicin/Bcl-2 grouping including the pro-apoptotic protein Bax, actinoporins derived from sea anemones, and the membrane attack complex-perforin/cholesterol dependent cytolysin (MACPF/CDC) set of proteins. We also explore how the structure and biological role of toroidal pores might be investigated further.
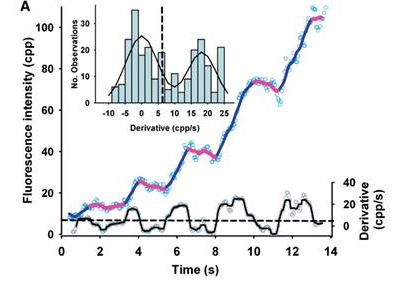
Photobleaching reveals heterogeneous stoichiometry for equinatoxin II oligomers.
10.1002/cbic.201300799
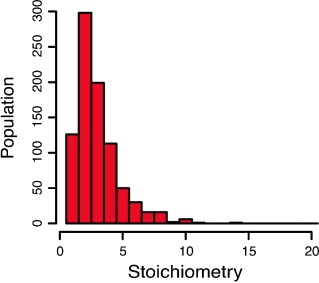
Single-molecule photobleaching of fluorescently labeled EqtII was used to determine the stoichiometry of EqtII oligomers in supported lipid bilayers.
Equinatoxin II (EqtII), a sea anemone cytolysin, is known to oligomerize to form pores that spontaneously insert into membranes. Crystallographic and cryo-EM studies of structurally similar cytolysins offer contradictory evidence for pore stoichiometry. Here we used single-molecule photobleaching of fluorescently labeled EqtII to determine the stoichiometry of EqtII oligomers in supported lipid bilayers. A frequency analysis of photobleaching steps revealed a log-normal distribution of stoichiometries with a mean of 3.4±2.3 standard deviations. Comparison of our experimental data with simulations of fixed stoichiometries supports our observation of a heterogeneous distribution of EqtII oligomerization. These data are consistent with a model of EqtII stoichiometry where pores are on average tetrameric, but with large variation in the number of subunits in individual pores.
High-speed single-particle tracking of GM1 in model membranes reveals anomalous diffusion due to interleaflet coupling and molecular pinning.
10.1021/nl502536u
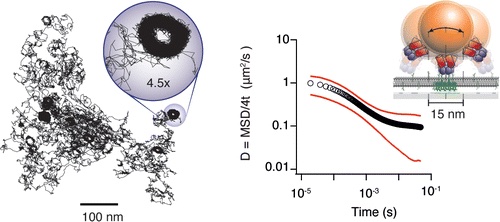
We use interferometric scattering microscopy to track molecules labeled with 20–40 nm scattering gold beads with simultaneous <2 nm spatial and 20 μs temporal precision.
The biological functions of the cell membrane are influenced by the mobility of its constituents, which are thought to be strongly affected by nanoscale structure and organization. Interactions with the actin cytoskeleton have been proposed as a potential mechanism with the control of mobility imparted through transmembrane “pickets” or GPI-anchored lipid nanodomains. This hypothesis is based on observations of molecular mobility using various methods, although many of these lack the spatiotemporal resolution required to fully capture all the details of the interaction dynamics. In addition, the validity of certain experimental approaches, particularly single-particle tracking, has been questioned due to a number of potential experimental artifacts. Here, we use interferometric scattering microscopy to track molecules labeled with 20-40 nm scattering gold beads with simultaneous 2 nm spatial and 20 μs temporal precision to investigate the existence and mechanistic origin of anomalous diffusion in bilayer membranes. We use supported lipid bilayers as a model system and demonstrate that the label does not influence time-dependent diffusion in the small particle limit (≤40 nm). By tracking the motion of the ganglioside lipid GM1 bound to the cholera toxin B subunit for different substrates and lipid tail properties, we show that molecular pinning and interleaflet coupling between lipid tail domains on a nanoscopic scale suffice to induce transient immobilization and thereby anomalous subdiffusion on the millisecond time scale.
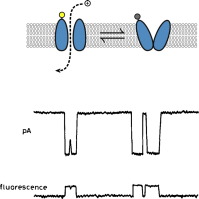
Simultaneous single-molecule fluorescence measurements of ion channel conformational change with single-channel electrophysiology.
Combining simultaneous single-molecule fluorescence measurements of ion channel conformational change with single-channel electrophysiology would enable a direct link between structure and function. Such methods would help us to create a truly molecular “movie” of how these important biomolecules work. Here we review past and recent progress toward this goal.
Fluorescence imaging of MACPF/CDC proteins. New techniques and their application.
10.1007/978-94-017-8881-6_15

This chapter introduces recent advances in fluorescence imaging that have the potential to overcome many of the obstacles that prevent a fuller understanding of MACPF/CDC proteins.
Structural and biochemical investigations have helped illuminate many of the important details of MACPF/CDC pore formation. However, conventional techniques are limited in their ability to tackle many of the remaining key questions, and new biophysical techniques might provide the means to improve our understanding. Here we attempt to identify the properties of MACPF/CDC proteins that warrant further study, and explore how new developments in fluorescence imaging are able to probe these properties.
Mass spectrometry defines the C-terminal dimerization domain and enables modeling of the structure of full-length OmpA.
10.1016/j.str.2014.03.004
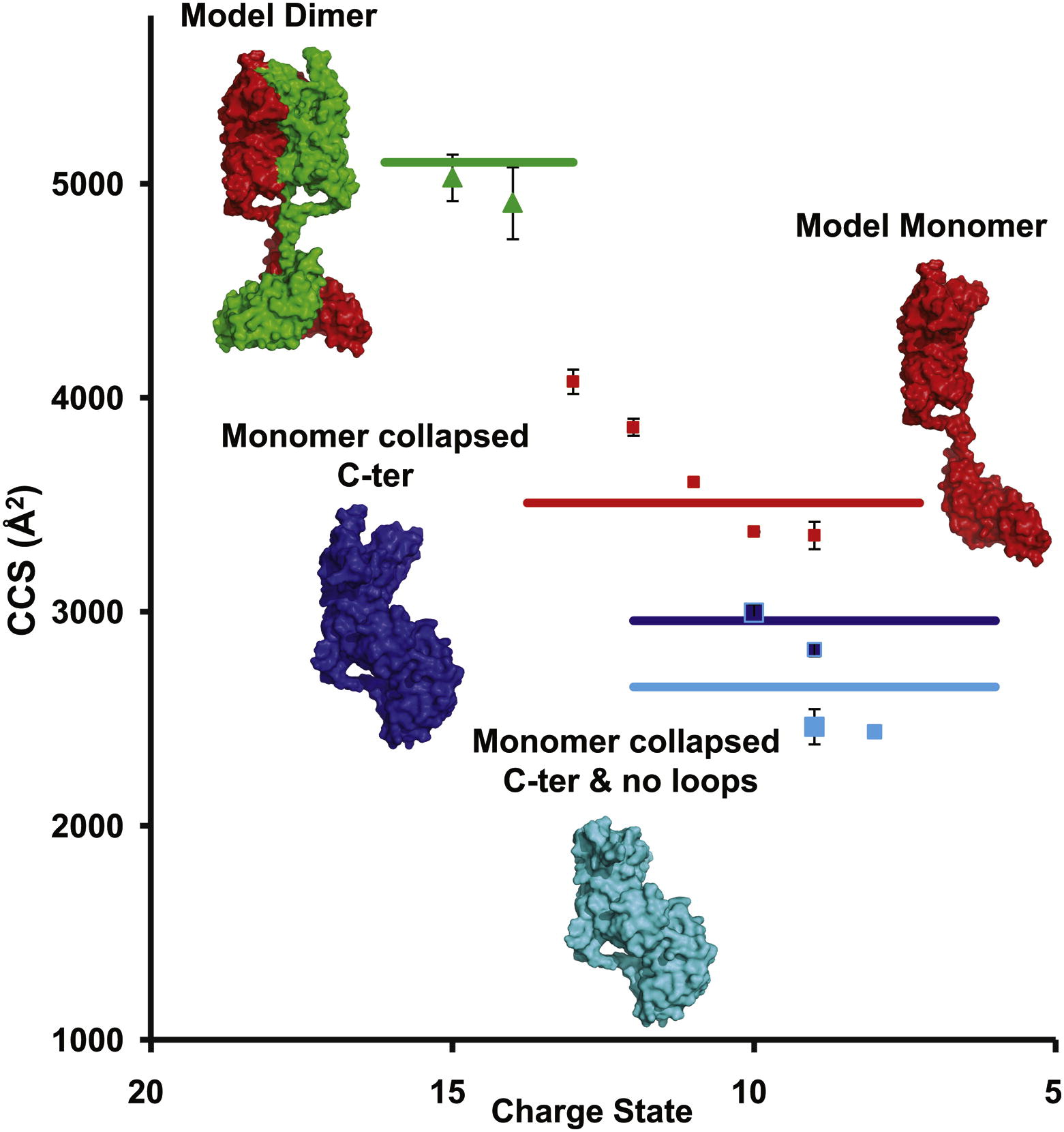
Full-length OmpA is partially present as a dimer.
The transmembrane domain of the outer membrane protein A (OmpA) from Escherichia coli is an excellent model for structural and folding studies of β-barrel membrane proteins. However, full-length OmpA resists crystallographic efforts, and the link between its function and tertiary structure remains controversial. Here we use site-directed mutagenesis and mass spectrometry of different constructs of OmpA, released in the gas phase from detergent micelles, to define the minimal region encompassing the C-terminal dimer interface. Combining knowledge of the location of the dimeric interface with molecular modeling and ion mobility data allows us to propose a low-resolution model for the full-length OmpA dimer. Our model of the dimer is in remarkable agreement with experimental ion mobility data, with none of the unfolding or collapse observed for full-length monomeric OmpA, implying that dimer formation stabilizes the overall structure and prevents collapse of the flexible linker that connects the two domains.
Imaging the lipid-phase-dependent pore formation of equinatoxin II in droplet interface bilayers.
10.1016/j.bpj.2013.11.4507
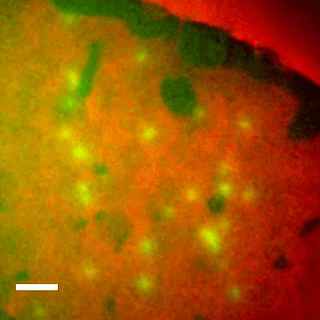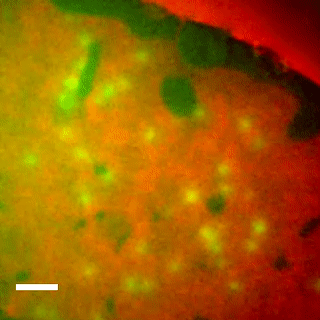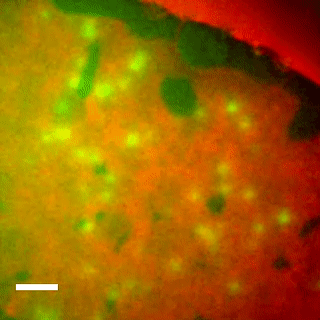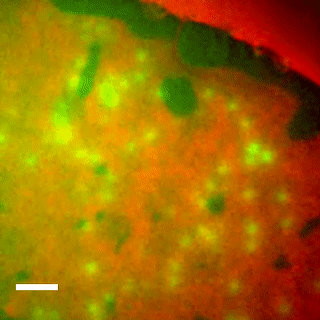
EqtII-Cy3B Pores Diffuse in the Ld Phase.
Using phase-separated droplet interface bilayers, we observe membrane binding and pore formation of a eukaryotic cytolysin, Equinatoxin II (EqtII). EqtII activity is known to depend on the presence of sphingomyelin in the target membrane and is enhanced by lipid phase separation. By imaging the ionic flux through individual pores in vitro, we observe that EqtII pores form predominantly within the liquid-disordered phase. We observe preferential binding of labeled EqtII at liquid-ordered/liquid-disordered domain boundaries before it accumulates in the liquid-disordered phase.
Live cell imaging shows reversible assembly of the TatA component of the twin-arginine protein transport system.
10.1073/pnas.1306738110
We used direct imaging of fluorescent protein-tagged Tat components in bacterial cells to show that the TatA element of the Tat system undergoes substrate- and proton motive force-dependent oligomerization.
The twin-arginine translocation (Tat) machinery transports folded proteins across the cytoplasmic membrane of bacteria and the thylakoid membrane of chloroplasts. It has been inferred that the Tat translocation site is assembled on demand by substrate-induced association of the protein TatA. We tested this model by imaging YFP-tagged TatA expressed at native levels in living Escherichia coli cells in the presence of low levels of the TatA paralogue TatE. Under these conditions the TatA-YFP fusion supports full physiological Tat transport activity. In agreement with the TatA association model, raising the number of transport-competent substrate proteins within the cell leads to an increase in the number of large TatA complexes present. Formation of these complexes requires both a functional TatBC substrate receptor and the transmembrane proton motive force (PMF). Removing the PMF causes TatA complexes to dissociate, except in strains with impaired Tat transport activity. Based on these observations we propose that TatA assembly reaches a critical point at which oligomerization can be reversed only by substrate transport. In contrast to TatA-YFP, the oligomeric states of TatB-YFP and TatC-YFP fusions are not affected by substrate or the PMF, although TatB-YFP oligomerization does require TatC.
A radical sense of direction. signalling and mechanism in cryptochrome magnetoreception.
10.1016/j.tibs.2013.07.002

Magnetoreception in many organisms is thought to involve cryptochrome proteins. A mechanism for this sensitivity is provided by the radical pair hypothesis.
The remarkable phenomenon of magnetoreception in migratory birds and other organisms has fascinated biologists for decades. Much evidence has accumulated to suggest that birds sense the magnetic field of the Earth using photochemical transformations in cryptochrome flavoproteins. In the last 5 years this highly interdisciplinary field has seen advances in structural biology, biophysics, spin chemistry, and genetic studies in model organisms. We review these developments and consider how this chemical signal can be integrated into the cellular response.
Constructing droplet interface bilayers from the contact of aqueous droplets in oil.
10.1038/nprot.2013.061

The basic rule of thumb is that if a lipid or solvent composition can be used for a conventional lipid bilayer experiment, it can be easily adapted to the DIB technique.
We describe a protocol for forming an artificial lipid bilayer by contacting nanoliter aqueous droplets in an oil solution in the presence of phospholipids. A lipid monolayer forms at each oil-water interface, and when two such monolayers touch, a bilayer is created. Droplet interface bilayers (DIBs) are a simple way to generate stable bilayers suitable for single-channel electrophysiology and optical imaging from a wide variety of preparations, ranging from purified proteins to reconstituted eukaryotic cell membrane fragments. Examples include purified proteins from the α-hemolysin pore from Staphylococcus aureus, the anthrax toxin pore and the 1.2-MDa mouse mechanosensitive channel MmPiezo1. Ion channels and ionotropic receptors can also be reconstituted from membrane fragments without further purification. We describe two approaches for forming DIBs. In one approach, a lipid bilayer is created between two aqueous droplets submerged in oil. In the other approach, a membrane is formed between an aqueous droplet and an agarose hydrogel, which allows imaging in addition to electrical recordings. The protocol takes approximately 30 min, including droplet generation, monolayer assembly and bilayer formation. In addition to the main protocol, we also describe the preparation of Ag/AgCl electrodes and sample preparation.
One-step synthesis of fluorescein modified nano-carbon for Pd(II) detection via fluorescence quenching.
10.1039/c2an16261j
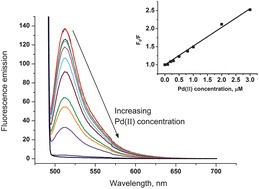
Carbon black nanoparticles modified with fluorescein.
Carbon black (CB) nanoparticles modified with fluorescein, a highly fluorescent molecule, were prepared using a facile and efficient methodology. Simply stirring CB in aqueous solution containing fluorescein resulted in the strong physisorption of fluorescein onto the CB surface. The resulting Fluorescein/CB was then characterised by means of X-ray photoelectron spectroscopy (XPS), cyclic voltammetry (CV), fluorescence microscopy and fluorescence spectroscopy. The optimum experimental conditions for fluorescence of Fluorescein/CB viz. fluorescence excitation and emission wavelengths, O(2) removal and the amount of Fluorescein/CB used, were investigated. The Fluorescein/CB was used as a fluorescent probe for the sensitive detection of Pd(II) in water, based on fluorescence quenching. The results demonstrated that the fluorescence intensity of Fluorescein/CB decreased with increasing Pd(II) concentration, and the fluorescence quenching process could be described by the Stern-Volmer equation. The limit of detection (LOD) for the fluorescence quenching of Fluorescein/CB by Pd(II) in aqueous solution was found to be 1.07 μM (based on 3σ). Last, approaches were studied for the removal of Fe(III) which interferes with the fluorescence quenching of Fluorescein/CB. Complexation of Fe(III) with salicylic acid was used to enhance and control the selectivity of Fluorescein/CB sensor towards Pd(II) in the presence of Fe(III).
Quantification of membrane protein inhibition by optical ion flux in a droplet interface bilayer array.
10.1002/anie.201107343

Droplet-based membrane protein screening.
Optical platforms for assaying membrane protein function offer a promising route to scalable high-throughput screening (see picture). For the first time quantitative measurements of membrane protein inhibition are reported in an optically addressable lipid bilayer array. Wide-field total internal reflection fluorescence (TIRF) imaging of Ca2+ flux enables the quantification of α-hemolysin inhibition by γ-cyclodextrin.

Rapid appearance of α-hemolysin oligomers.
We have observed the assembly of the staphylococcal pore-forming toxin α-hemolysin using single-molecule fluorescence imaging. Surprisingly, assembly from the monomer to the complete heptamer is extremely rapid, occurring in less than 5 ms. No lower order oligomeric intermediates are detected. Monte Carlo simulation of our experiment shows that assembly is diffusion limited, and pore formation is dependent on the stability of intermediate species. There are close similarities between bacterial pore-forming toxins, such as staphylococcal α-hemolysin, the anthrax protective antigen, and the cholesterol-dependent cytolysins, and their eukaryotic analogs, such as the complement pore membrane attack complex and perforin domain. The assembly mechanism we have observed for α-hemolysin provides a simple model that aids our understanding of these important pore formers.
Determining membrane capacitance by dynamic control of droplet interface bilayer area.
10.1021/la203081v
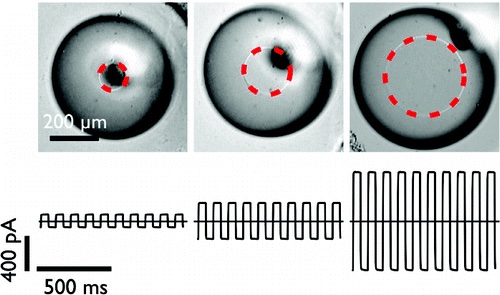
Dynamic control of droplet interface bilayer area.
By making dynamic changes to the area of a droplet interface bilayer (DIB), we are able to measure the specific capacitance of lipid bilayers with improved accuracy and precision over existing methods. The dependence of membrane specific capacitance on the chain-length of the alkane oil present in the bilayer is similar to that observed in black lipid membranes. In contrast to conventional artificial bilayers, DIBs are not confined by an aperture, which enables us to determine that the dependence of whole bilayer capacitance on applied potential is predominantly a result of a spontaneous increase in bilayer area. This area change arises from the creation of new bilayer at the three phase interface and is driven by changes in surface tension with applied potential that can be described by the Young-Lippmann equation. By accounting for this area change, we are able to determine the proportion of the capacitance dependence that arises from a change in specific capacitance with applied potential. This method provides a new tool with which to investigate the vertical compression of the bilayer and understand the changes in bilayer thickness with applied potential. We find that, for 1,2-diphytanoyl-sn-glycero-3-phosphocholine membranes in hexadecane, specific bilayer capacitance varies by 0.6-1.5% over an applied potential of ±100 mV.
Alamethicin.
Alamethicin is the archetypal antimicrobial pore-forming peptide. Although the peptide has long been known to form pores of characteristic conductances in lipid membranes, the precise nature of these pores is not known. Simultaneous calcium-flux imaging and single-channel recording in a droplet interface bilayer allowed us to directly attribute multiple conductance states to a single point diffusing in the bilayer.
Dynamic and reversible control of 2D membrane protein concentration in a droplet interface bilayer.
10.1021/nl201689v

Controlling membrane protein surface density.
We form an artificial lipid bilayer between a nanolitre aqueous droplet and a supporting hydrogel immersed in an oil/lipid solution. Manipulation of the axial position of the droplet relative to the hydrogel controls the size of the bilayer formed at the interface; this enables the surface density of integral membrane proteins to be controlled. We are able to modulate the surface density of the β-barrel pore-forming toxin α-hemolysin over a range of 4 orders of magnitude within a time frame of a few seconds. The concentration changes are fully reversible. Membrane protein function and diffusion are unaltered, as measured by single molecule microscopy and single channel electrical recording.
In vitro reconstitution of eukaryotic ion channels using droplet interface bilayers.
10.1021/ja200128n
Routine reconstitution of membrane proteins.
The ability to routinely study eukaryotic ion channels in a synthetic lipid environment would have a major impact on our understanding of how different lipids influence ion channel function. Here, we describe a straightforward, detergent-free method for the in vitro reconstitution of eukaryotic ion channels and ionotropic receptors into droplet interface bilayers and measure their electrical activity at both the macroscopic and single-channel level. We explore the general applicability of this method by reconstitution of channels from a wide range of sources including recombinant cell lines and native tissues, as well as preparations that are difficult to study by conventional methods including erythrocytes and mitochondria.
DNA unwinding by AddAB is characterized by pauses and bursts of activity.
DNA helicases are motor proteins that catalyze the unwinding of double-stranded DNA into single-stranded DNA using the free energy from ATP hydrolysis. Single molecule approaches enable us to address detailed mechanistic questions about how such enzymes move processively along DNA. Here, an optical method has been developed to follow the unwinding of multiple DNA molecules simultaneously in real time. This was achieved by measuring the accumulation of fluorescent single-stranded DNA-binding protein on the single-stranded DNA product of the helicase, using total internal reflection fluorescence microscopy. By immobilizing either the DNA or helicase, localized increase in fluorescence provides information about the rate of unwinding and the processivity of individual enzymes. In addition, it reveals details of the unwinding process, such as pauses and bursts of activity. The generic and versatile nature of the assay makes it applicable to a variety of DNA helicases and DNA templates. The method is an important addition to the single-molecule toolbox available for studying DNA processing enzymes.
Lucky imaging. improved localization accuracy for single molecule imaging.
10.1016/j.bpj.2008.12.3945
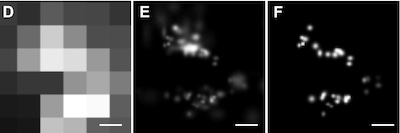
Lucky imaging improves superresolution.
We apply the astronomical data-analysis technique, Lucky imaging, to improve resolution in single molecule fluorescence microscopy. We show that by selectively discarding data points from individual single-molecule trajectories, imaging resolution can be improved by a factor of 1.6 for individual fluorophores and up to 5.6 for more complex images. The method is illustrated using images of fluorescent dye molecules and quantum dots, and the in vivo imaging of fluorescently labeled linker for activation of T cells.
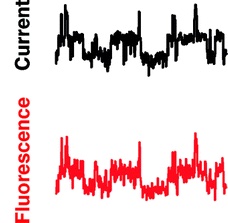
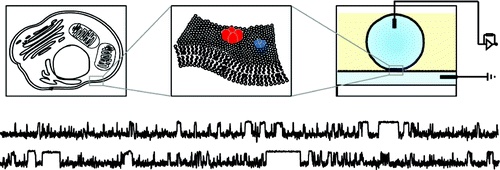
Simultaneous measurement of ionic current and fluorescence from single protein pores.
10.1021/ja808128s

Simultaneous fluorescence and electrical detection of stochastic blocking.
The ability to simultaneously monitor both the ionic current and fluorescence from membrane channels and pores has the potential to link structural changes with function in such proteins. We present a new method for simultaneously measuring single-channel electrical currents and fluorescence from membrane proteins by using water-in-oil droplet bilayers. We demonstrate the simultaneous fluorescence and electrical detection of stochastic blocking by cyclodextrin in multiple staphylococcal alpha-hemolysin pores. The combined fluorescence signal from individual pores exhibits the same sequence of blocking events as the total current recording, showing that the two signals from each pore are correlated.

Our Chapter in Single Molecule Biology by Alex Knight.
Single molecule fluorescence (SMF) methods have made major contributions to the current understanding of membrane biology. This chapter discusses findings obtained by SMF into the mechanisms that govern membrane structure, cell signaling, ion channel gating, and vesicle transport. It also considers how recent advances in single molecule imaging technology could be applied to membranes and used to identify technological limitations currently inhibiting the field. Several key characteristics of membranes and their components make them well suited to single molecule approaches, including their accessibility to in vivo studies due to their location at the cell surface, the availability of model cell membranes for in vitro studies, and the confinement of molecules to 2D motion. SMF imaging of membranes in vivo requires the specific attachment of fluorescent labels to proteins or lipids of interest within the membrane of live cells, and the primary challenges facing membrane studies are related to labeling and detection technology.
Single molecule methods have emerged as a powerful new tool for exploring biological phenomena. We provide a brief overview of the scope of current experiments and assess the limitations of both fluorescent labels and the means to achieve protein modification for single molecule microscopy.

Droplet Interface Bilayer.
Our efforts on droplet interface bilayers in Oxford began in the summer of 2005 following a suggestion made by David Needham at “Lipids, liposomes and biomembranes”, a conference in Vancouver. He proposed that a bilayer would form between two droplets brought together in an oil (squalene) containing a lipid (glycerol monooleate) and that this might be a means to miniaturize planar bilayer recording.
Direct detection of membrane channels from gels using water-in-oil droplet bilayers.
10.1021/ja075715hS
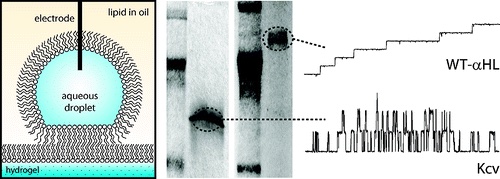
Detecting ion channels directly from gel electrophoresis.
We form planar lipid bilayers between an aqueous droplet and a hydrogel support immersed in a lipid-oil solution. By scanning the bilayer over the surface of an SDS-PAGE gel, we are able to directly detect membrane proteins from gels using single-channel recording. Using this technique, we are able to examine low levels of endogenous protein from cell extracts without the need for over-expression. We also use droplet bilayers to detect small molecules from hydrogels. The bilayers show enhanced stability compared to conventional planar lipid bilayers, and both bilayer size and position can be controlled during an experiment. Hydrogel scanning with droplet bilayers provides a new method for the discovery and characterization of ion channels with the potential for high-throughput screening.
Enhanced stability and fluidity in droplet on hydrogel bilayers for measuring membrane protein diffusion.
10.1021/nl071943y
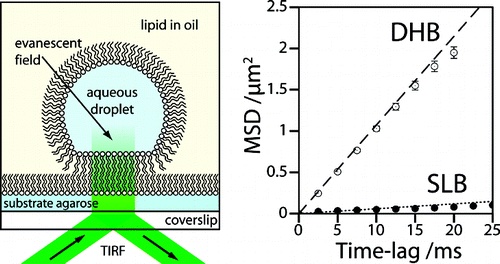
DIBs are good for membrane proteins.
We form artificial lipid bilayers suitable for single-molecule fluorescence microscopy by contacting an aqueous droplet with a hydrogel support immersed in a solution of lipid in oil. Our results show that droplet on hydrogel bilayers (DHBs) have high lipid mobilities, similar to those observed in unsupported lipid bilayers. DHBs are also stable over a period of several weeks. We examine membrane protein diffusion in these bilayers and report a decreased lateral mobility of the heptameric beta-barrel pore-forming toxin alpha-hemolysin versus that of its monomeric precursor. These results corroborate previous models of the alpha-hemolysin insertion mechanism where the monomer binds to the lipid bilayer without insertion.
Membrane protein stoichiometry determined from the step-wise photobleaching of dye-labelled subunits.
10.1002/cbic.200600474
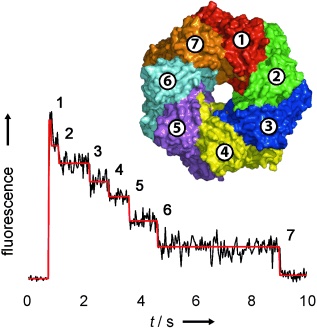
Counting membrane protein stoichiometry.
In a generally applicable approach, the number of subunits in fluorescently-labelled protein complexes has been determined by counting photobleaching steps from individual molecules (see figure). The distribution of steps in the pore-forming toxins α-haemolysin and leukocidin indicate seven subunits for α-hemolysin, and four LukF and four LukS subunits for leukocidin.
A key to understanding bacterial pathogenicity is the mechanism by which water-soluble protein toxins assemble on cell membranes to form oligomeric bilayer-spanning pores. The recent reconstructions from cryo-electron micrographs of three-dimensional pore and prepore structures of the cholesterol-dependent toxin pneumolysin shed new light on the later steps of the assembly of large toxin pores.
A model of stereocilia adaptation based on single molecule mechanical studies of myosin I.
10.1098/rstb.2004.1559
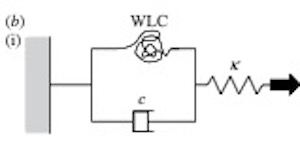
Worm-like chains explain sterocilia myosin adaptation.
We have used an optical tweezers-based apparatus to perform single molecule mechanical experiments using the unconventional myosins, Myo1b and Myo1c. The single-headed nature and slow ATPase kinetics of these myosins make them ideal for detailed studies of the molecular mechanism of force generation by acto-myosin. Myo1c exhibits several features that have not been seen using fast skeletal muscle myosin II. (i) The working stroke occurs in two, distinct phases, producing an initial 3 nm and then a further 1.5 nm of movement. (ii) Two types of binding interaction were observed: short-lived ATP-independent binding events that produced no movement and longer-lived, ATP-dependent events that produced a full working stroke. The stiffness of both types of interaction was similar. (iii) In a new type of experiment, using feedback to apply controlled displacements to a single acto-myosin cross-bridge, we found abrupt changes in force during attachment of the acto-Myo1b cross-bridge, a result that is consistent with the classical ‘T2’ behaviour of single muscle fibres. Given that these myosins might exhibit the classical T2 behaviour, we propose a new model to explain the slow phase of sensory adaptation of the hair cells of the inner ear.
![]()
Linking optical tweezers to super-resolved tracking.
The movement produced by a small number of myosin molecular motors was measured with nanometre precision using single-molecule fluorescence localisation methods. The positional precision of the measurements was sufficient to reveal fluctuations in sliding velocity due to stochastic interactions between individual myosin motors and the actin filament. Dependence of sliding velocity upon filament length was measured and fluctuations in velocity were quantified by autocorrelation analysis. Optical tweezers-based nanometry was used to measure the myosin-1b step-size directly. The 10 nm power-stroke and its duty cycle ratio were consistent with values derived from in vitro sliding assays.
An advance that counts! A method is presented for analyzing single-molecule behavior using one-photon excitation of fluorescence. It is based on the photon counting histogram (PCH) procedure that has enjoyed much success for two-photon excitation, but has not previously been applied to one-photon excitation. This corrected PCH model is able to resolve fluorescent species with different degrees of brightness, for example, a mixture of tetramethylrhodamine-5′-maleimide and Cy3-maleimide (see graph). Because one-photon excitation is often much easier to do than two-photon excitation, this method has the potential to be widely used and represents an important advance in the field of single-molecule spectroscopy.
Recent advances in single-molecule techniques allow the application of force to an individual biomolecule whilst simultaneously monitoring its response using fluorescent probes. The effects of applied mechanical load on single-enzyme turnovers, biomolecular interactions and conformational changes can now be studied with nanometer precision and millisecond time resolution.
Coupled electrorotation of polymer microspheres for microfluidic sensing and mixing.
10.1021/ac0258599
Coupling between microscale objects in an AC field causes rotation and mixing.
We show that coupled electrorotation (CER) of microscopic particles using microfabricated electrodes can be used for localized sensing and mixing. The effective use of microelectromechanical systems and micro total analysis systems requires many types of control. These include the abilityto (1) manipulate objects within microchannels by noncontact means, (2) mix fluids, and (3) sense local chemical parameters. Coupled electrorotation, in which the interactions between induced electric dipoles of adjacent particles lead to particle rotation, addresses aspects of all three challenges simultaneously. CER is a simple means of controlling the rotation of dielectric objects using homogeneous external radio frequency electric fields. CER is sensitive to several chemical and physical parameters such as the solution conductivity, pH, and viscosity. As a step toward integrating CER devices into microfluidic systems, a simple chip was designed to induce local mixing and to detect local changes in salt concentration, pH, and viscosity.
Combining FRET and FCS to understand DNA conformational dynamics.
Intramolecular chain diffusion is an elementary process in the conformational fluctuations of the DNA hairpin-loop. We have studied the temperature and viscosity dependence of a model DNA hairpin-loop by FRET (fluorescence resonance energy transfer) fluctuation spectroscopy (FRETfs). Apparent thermodynamic parameters were obtained by analyzing the correlation amplitude through a two-state model and are consistent with steady-state fluorescence measurements. The kinetics of closing the loop show non-Arrhenius behavior, in agreement with theoretical prediction and other experimental measurements on peptide folding. The fluctuation rates show a fractional power dependence (beta = 0.83) on the solution viscosity. A much slower intrachain diffusion coefficient in comparison to that of polypeptides was derived based on the first passage time theory of SSS [Szabo, A., Schulten, K. and Schulten, Z. (1980) J. Chem. Phys. 72, 4350-4357], suggesting that intrachain interactions, especially stacking interaction in the loop, might increase the roughness of the free energy surface of the DNA hairpin-loop.
Stretched exponential kinetics have been observed in the conformational fluctuation of a DNA hairpin-loop under equilibrium conditions. In this paper, we employ a simple multiple-pathway two-state jump model to calculate single-molecule proximity ratio distributions. The simulation can reasonably reproduce the experimental single-molecule data of the conformational fluctuations in water, indicating that static disorder is dominant. In contrast, there exists significant discrepancy between the two-state simulation and experiment in buffer (2.5 mM Tris-HCl, 250 lM EDTA, 100 mM NaCl), suggesting that both static and dynamic disorder may contribute to the non-exponential kinetics.
Diamond deposition in a DC-arc Jet CVD system. investigations of the effects of nitrogen addition.
10.1016/S0925-9635(00)00444-1
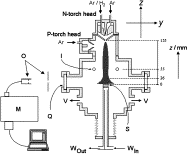
Making Diamond with a satellite thruster.
Studies of the chemical vapour deposition of diamond films at growth rates >100 um h-1 with a 10-kW DC-arc jet system are described. Additions of small amounts of N2 to the standard CH4/H2/Ar feedstock gas results in strong CN(B->X). emission, and quenches C2(d->a). and H-alpha emissions from the plasma. Species selective, spatially resolved optical emission measurements have enabled derivation of the longitudinal and lateral variation of emitting C2, CN radicals and H (n=3) atoms within the plasma jet. Scanning electron microscopy and laser Raman analyses indicate that N2 additions also degrade both the growth rate and quality of the deposited diamond film; the latter technique also provides some evidence for nitrogen inclusion within the films.
darm.gif
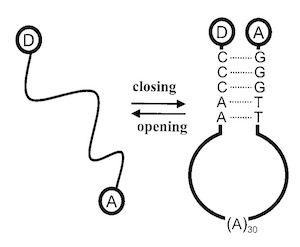
FRET fluctuation spectroscopy. Exploring the conformational dynamics of a DNA hairpin-loop.
10.1021/jp001560n
The motions of a dye-labeled DNA hairpin loop (Cy5-5′-GGGTT-(A)30-AACCC-3′-TMR) have been investigated through the fluctuations in proximity ratio from fluorescence resonance energy transfer (FRET). We examine three solution conditions: (1) MilliQ water, (2) Tris-EDTA buffer, and (3) Tris-EDTA buffer plus an excess of DNA complementary to the loop sequence, (T)30. Correlations in proximity ratio show submillisecond dynamics. Static heterogeneity is revealed from the distribution of proximity ratio amplitudes. The observed stretched exponential kinetics are consistent with a model based on the transition between two states over a complex energy landscape.
Ratiometric analysis of single-molecule Fluorescence Resonance Energy Transfer using logical combinations of threshold criteria. A study of 12-mer DNA.
10.1021/jp993914k
Single-molecule fluorescence resonance energy transfer (FRET) combined with bulk fluorescence lifetimes, anisotropy, and spectra have been used to study a donor-acceptor labeled model DNA system (Cy5-5′- ACCTGCCGACGC-3′-TMR). A general ratiometric analysis method using independent donor and acceptor thresholding has been developed. Use of two logical combinations of thresholding criteria provides more information than either method alone, revealing heterogeneity within this system. Conditions yielding similar bulk fluorescence spectra can be readily distinguished by this single-molecule method. Fluorescence lifetimes and anisotropy measurements also suggest nonnegligible fluorophore-DNA interaction.